
Translate this page into:
Myositis ossificans progressiva: A clinico-radiological evaluation-Case report with brief review of literature
Address for correspondence: Prof. Prashant K. Gupta, 108, Chanakyapuri, Shastri Nagar, Meerut - 250 004, Uttar Pradesh, India. E-mail: prashantk.gupta@yahoo.com
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
How to cite this article: Rathee N, Gupta PK, Gupta K, Garg G. Myositis ossificans progressiva: A clinico-radiological evaluation- Case report with brief review of literature. J Orthop Allied Sci 2016;4:87-90.
Abstract
Myositis ossificans progressiva/fibrodysplasia ossificans progressiva (MOP), is an autosomal dominant mesodermal tissue disorder, characterized by an initial period of inflammation and subsequent proliferation of fibrous tissue with the formation of ectopic bone tissue. The incidence of MOP is one case per two million people. The ectopic bone tissue formed is located in soft tissue mainly in the connective tissue of striated musculature. We report MOP in an 18-year old female who presented with multiple tender, hard swelling in various parts of the body associated with stiffness and limitations of movements. A literature review of the subject showed few similar case reports in the literature. We revisit the criteria for diagnosis and the essentials of management and treatment of MOP as it is rare being a rare condition, and treatment guidelines are not clear.
Keywords
Bone
heterotopic
myositis

Introduction
Myositis ossificans progressiva (MOP) is a rare, autosomal dominant[1,2,3] disease affecting all ethnic backgrounds.[4] It is estimated that the incidence of the disease is 1 per 2 million people, and the prevalence is 2500 cases worldwide.[5] It is a connective tissue disease characterized by widespread, progressive, ectopic ossification of soft tissues (striated muscles, tendons, fasciae, ligaments, and subcutaneous tissues).[6] It is particularly disabling in children and is characterized by two cardinal features: heterotopic progressive osteogenesis and congenital abnormalities of the great toes.[7,8] Classically, the disease is characterized by heterotopic ossification of soft tissues, which is usually complicated by restriction of movements at the affected sites. It usually starts and progresses in a craniocaudal, dorsal-ventral and proximodistal manner.[9] However, flares of the disease might occur at sites of trauma or injury.[10] The tongue, smooth muscles, diaphragmatic muscles and cardiac muscles are fortunately spared.[11]
The term fibrodysplasia ossificans progressiva is preferred to myositis ossificans because ectopic osteogenesis occurs in the connective tissue within muscles, fasciae, ligaments, tendons, and joint capsules, rather than in the muscle fibers themselves. These may show nonspecific, possibly secondary pathological changes.[8] There is no treatment for the Munchmeyer's disease.[12] However, knowledge of the genetic mutation will now provide hope for drug development and gene therapy for these patient's misery.[13] Since curative therapy is not available, management is based on the principle of primum non-nocere, particularly at preventing abnormal ossification. Therefore, an increased awareness of the disease among clinicians is of great importance.
We report on a patient with MOP having the progressive restriction of multiple joint movements.
Case Report
The patient was an 18-year-old female who was referred to radiology department with multiple bony hard swellings with scoliosis causing limitation of movements and physical deformities. On further questioning, she was noted by her parents to have deformities and shortening of the great toes since childhood. The patient's past medical history revealed multiple episodes of spontaneous swellings at multiple sites, including chin, back, right upper limb and lower limbs. There was progressive restriction of movements of the hips, neck, right upper limb and lumbar spine with noticeable deformities. There was no familial relationship between the parents, and none of his family members had a similar problem. On examination, the patient was noted to have multiple nontender firm subcutaneous nodules in the chin, neck, and back with scoliosis of the lumbar spine. Both the great toes were short with valgus deformity [Figure 1].
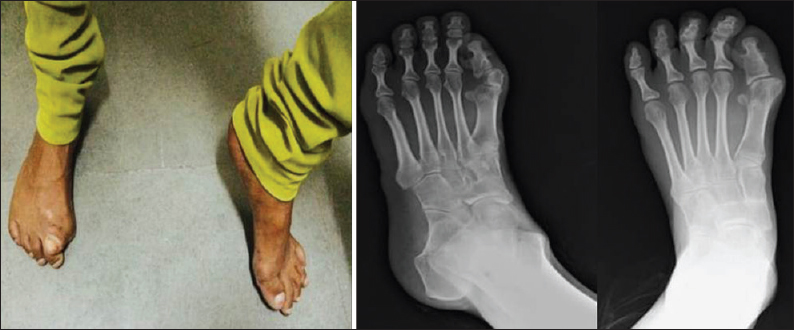
- X-ray and photograph of both foot showed great toes were short with valgus deformity
The radiological evaluation showed expansion of the posterior column of cervical vertebrae, as well as calcification and bony fusion with an ossified bridge between chin and sternum with straightening of the cervical spine [Figure 2]. The lumbosacral spine showed severe scoliosis with heterotopic ossification along the soft tissues dorsally [Figure 3], and the pelvis showed ossified bridges around the hip joints, extending from the ischium to the greater and lesser trochanters and the proximal shaft of femur [Figure 4]. Radiograph AP view of thigh and leg showing multifocal heterotopic ossification [Figures 5 and 6]. The thorax showed extensive multifocal heterotopic ossification and osseous bridging between ribs and ossified bridging of the right humerus [Figure 7] Blood and serological studies showed a normal hemogram, erythrocyte sedimentation rate, and serum calcium, alkaline phosphatase, creatine phosphokinase, alanine and aspartate transaminases, routine urinalysis, and creatinine clearance were within normal limits.
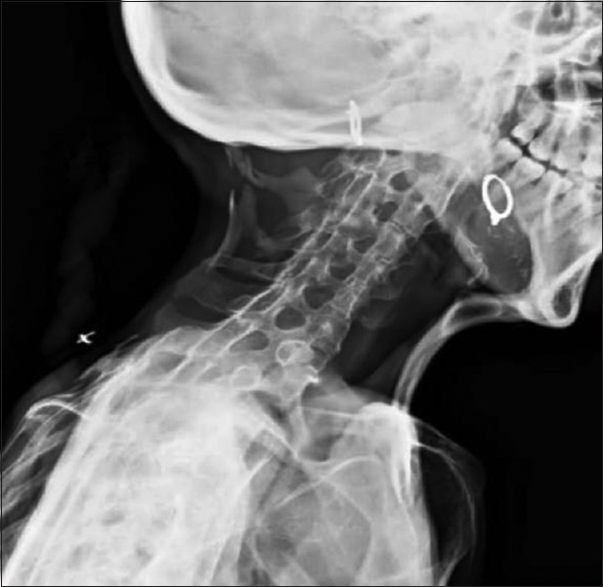
- The cervical spine showed expansion of the posterior column of cervical vertebrae with calcification and bony fusion and ossified bridge between chin and sternum
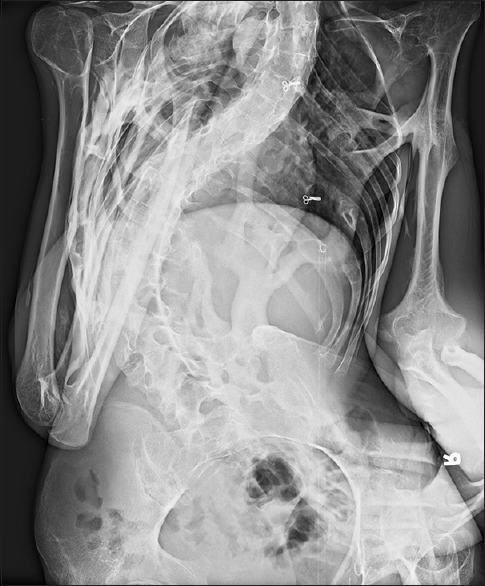
- Lumbosacral spine showed severe scoliosis with heterotopic ossification of soft tissues dorsally
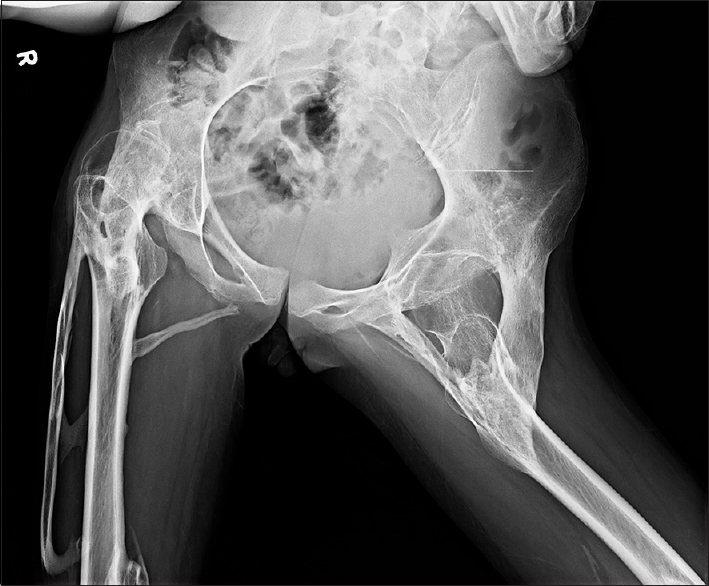
- Pelvis showed ossified bridges around the hip joints, extending from the ischium to the greater and lesser trochanters and to the proximal shaft of femur
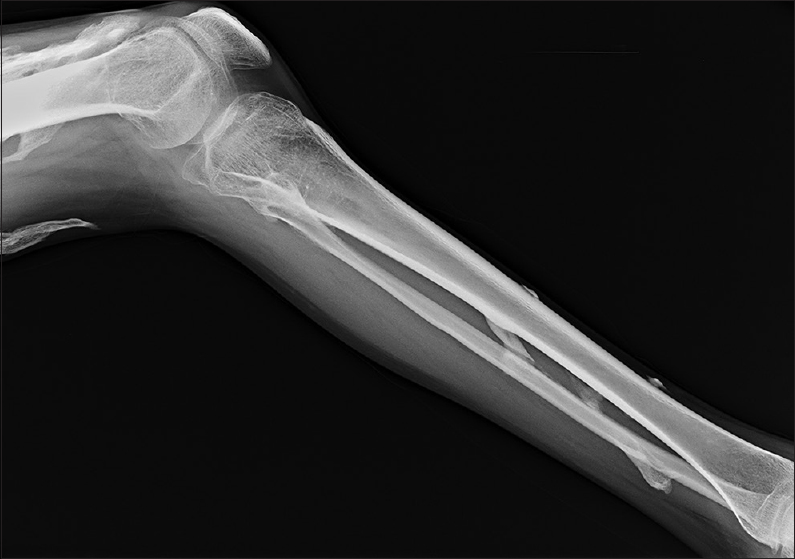
- Radiograph of leg showing multifocal heterotopic ossification
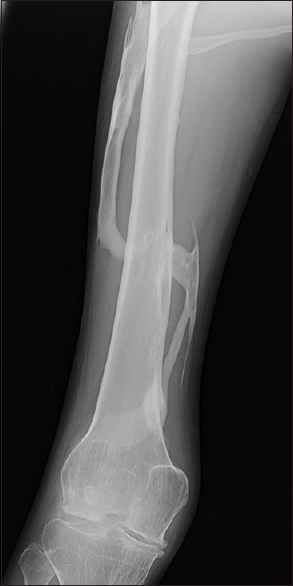
- Radiograph of thigh showing multifocal heterotopic ossification
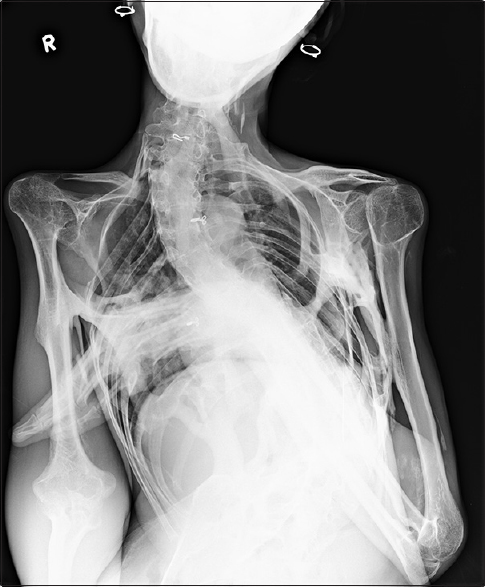
- Thorax showed extensive multifocal heterotopic ossification and osseous bridging between ribs and ossified bridging of the right humerus
Discussion
MOP is a rare autosomal dominant disease which should be diagnosed noninvasively as early as possible, based on history, clinical, and radiological findings.[7] It is clinically characterized by two main features, anomalies of the great toes and thumbs and progressive ectopic ossification of soft tissues causing limitation of movements.
Although the average onset age of the disease is about 5 years,[14] the affected child might be noticed after birth to have short and deformed great toes which occur in most of the cases of MOF. This was noticed by the parents of the patient during childhood. From birth until adolescence, the patient experiences spontaneous hard soft tissue swellings, which are usually painful.[10] These symptoms are usually the initial presentation of the patient and there might be flare-ups after trauma. Some cases present intensely with acute torticollis and painful mass in the sternocleidomastoid.[14] In most cases, it starts at the neck progressing downward to affect the thoracic and lumbar regions, and then the limbs.[10] Sternocleidomastoid is often the initial site of involvement, progresses to shoulder girdle, upper arms, spine and pelvis. The end result is bridging between extremities and torso, between ribs and between thorax and pelvis with severe restriction of motion.[14] As it was the case in our patient, scoliosis is the end result of this heterotopic ossification that occurs more on one side.[9] Even though the lungs are not usually affected directly by the disease, recurrent infections, and atelectasis usually occur due to restriction of chest expansion secondary to disease progression.[15] Affected patients usually become dependent and confined to wheelchair or bed at the second decade of their life as a result of ankylosing of all major joints of both axial and appendicular skeleton. Many patients were reported to have conductive hearing loss due to fusion of the ear ossicles.[10]
MOP is generally a clinical diagnosis that is based on the presence of congenital anomalies of the great toes, progressive heterotopic ossification, and the classical pattern of disease progression. Biopsies are not recommended for the diagnosis because they might worsen the ossification at the site, and diagnosis might be confused with bone malignancies like fibrosarcoma.[16]
Routine laboratory tests including calcemia and phosphatemia are usually normal or noncontributory in MOP. Roentgenograms may aid in documenting minor osseous dysmorphism. Bone scintigraphy with99m Tc-methylene diphosphonate may demonstrate early the heterotopic ossification and aid in the assessment of the extent and progression of the disease.
Conclusion
Early diagnosis prevents catastrophic harmful diagnostic and treatment procedures. It usually presents in a classical pattern with characteristic radiological findings on plain films. There is no effective treatment that can cure it or stop its progression. Physicians, health-care professionals, patients, and their families must be educated about the disease. Although drugs can be used to decrease some symptoms, the best approach is still the early diagnosis and prevention of trauma that can provide a better quality of life.
Financial support and sponsorship
Nil
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- A three generation family with fibrodysplasia ossificans progressiva. J Med Genet. 1993;30:687-9.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am. 1993;75:1214-20.
- [CrossRef] [PubMed] [Google Scholar]
- Mild expression of fibrodysplasia ossificans progressiva: A report of 3 cases. J Rheumatol. 1995;22:976-8.
- [Google Scholar]
- The genetics of fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1998;346:15-8.
- [CrossRef] [Google Scholar]
- Fibrodysplasia (myositis) ossificans progressiva. Clinical lessons from a rare disease. Clin Orthop Relat Res. 1998;346:7-14.
- [CrossRef] [Google Scholar]
- Fibrodysplasia ossificans progressiva: Case report. Arq Neuropsiquiatr. 2000;58:342-7.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrodysplasia ossificans progressiva: Still turning into wood after 300 years? Eur J Pediatr. 1995;154:694-9.
- [CrossRef] [PubMed] [Google Scholar]
- Myositis ossificans progressiva. In: Brooke MH, ed. A Clinicians View of Neuromuscular Diseases (2nd). Baltimore: Williams & Wilkins; 1986. p. :239-42.
- [Google Scholar]
- Unusual presentations in myositis ossificans progressiva. A case report. Acta Orthop Belg. 2001;67:86-9.
- [Google Scholar]
- Fibrodysplasia ossificans progressiva: Case report. Arq Neuropsiquiatr. 2005;63:1090-3.
- [CrossRef] [PubMed] [Google Scholar]
- Myositis ossificans & fibrodysplasia ossificans progressiva. In: Taveras J, Ferrucci JT, eds. Radiology: Diagnosis, Imaging, Intervention. Vol 5. Philadelphia: Lippincott Raven Publishers; 1996. p. :6-9. Ch. 132
- [Google Scholar]
- Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:e654-61.
- [CrossRef] [PubMed] [Google Scholar]
- A new era for fibrodysplasia ossificans progressiva: A druggable target for the second skeleton. Expert Opin Biol Ther. 2007;7:705-12.
- [CrossRef] [PubMed] [Google Scholar]
- Contrast-enhanced MRI of an early preosseous lesion of fibrodysplasia ossificans progressiva in a 21-month-old boy. AJR Am J Roentgenol. 2003;181:1145-7.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary and cardiac function in advanced fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1998;346:104-9.
- [CrossRef] [Google Scholar]
- Fibrodysplasia ossificans progressiva. AJR Am J Roentgenol. 1982;139:935-41.
- [CrossRef] [PubMed] [Google Scholar]




