
Translate this page into:
Klippel–Trénaunay Syndrome (KTS or KT) is a rare clinical syndrome
Address for correspondence: Dr. Jitendra Aloria, Govt. Medical College & Attached Groups of Hospitals, Arya Hospitals near police line Sikar, Rajasthan, 332001, India. E-mail: jitendra.170@gmail.com
-
Received: ,
Accepted: ,
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
How to cite this article: Gosaliya MK, Aloria J, Goel R, Bairwa DK, Maheshwari M, Pachori S. Klippel–Trénaunay Syndrome (KTS or KT) is a rare clinical syndrome. J Orthop Spine 2022;10:40-3.
Abstract
Klippel Trenaunay syndrome is a congenital illness that affects capillary abnormalities, varicosities, and musculoskeletal hypertrophy. That condition incorporates a range of pathology, comprising haemorrhage, venous thromboembolism, embolic consequences, with, in exceptional situations, appendage elongation, which may necessitate surgery. Venous aberrations are divided into the following but never pass the centreline. Nevertheless, we encountered a scenario of an 8-year-old kid that manifested having varicosity of vasculature or deformation of the right lower extremity, as well as cavernous haemangiomas distributed everywhere in his chest, back, gluteal area, and legs since new. Due to the involvement of neurofibromatosis, several paravertebral soft tissue masses and bladder hypertrophy were also seen. In clinical practise, the coexistence of KTS with neurofibromatosis is uncommon.
Keywords
Capillary abnormalities
compression stockings
Klippel–Trénaunay syndrome
port-wine stains
soft tissue and/or bone hypertrophy
venous varicosity

Introduction
Klippel–Trénaunay syndrome (KTS or KT) is a rare dermatological condition characterised by delicate tissue or bone overgrowths, dermatological vascular abnormalities, and venous capillary and lymphatic malformations. Cavernous lymphatic venous malformation (KTS) is the new term for KTS (CLVM). An arteriovenous malformation, on the other hand, is distinguished by a high flow rate. Klippel-Tranquille-Weber syndrome, on the other hand, is a separate disorder. The vascular abnormalities are usually only visible in one limb, but in rare cases, many limbs might well be afflicted. Lymphocytic filariasis, Beckwith–Wiedemann syndromes, Proteus disease, Russell–Silver disorder, Maffucci disorder, congenital hemidysplasia involving ichthyosiform erythroderma or extremity impairments), neurofibromatosis type 1 and triploid syndrome should all be thoroughly and promptly evaluated. When it comes to long-term follow-up, management is frequently hesitant.1st
Case Report
An 8-year-old boy approached us with characteristics of subepithelial plaques across his right lower extremities, involving ulceration and bleeding after light trauma in specific locations just above the plaques. In concluding, this client displayed several fluid-filled lesions interspersed with subepithelial pustules and plaques, along with right lower extremity expansion. It exhibited severe anaemia (Hb8mg/dL) according to the complete physical assessment. Their heart rate was normal at 90 beats per minute, and their blood pressure in the right upper arm was 110/68mmHg. He had a normal temperature and no signs of jaundice, cyanosis, clubbing, or lymph node hypertrophy. His right lower limb had numerous hypertrophies. Detailed examination revealed several discretered to blueish black papulonodular angiokeratomas present over the port-winestain across the majority of the thematical side of the right lower leg [Figure 1]. Similar lesions were also discovered on the lower back and buttocks [Figure 2]. There were numerous hypertrophies over the right knee joint. The perianal region is riddled with lymphangiectasis lesions. When both lower limbs were measured, it was discovered that the right lower limb was 5.8 cm longer than the left lower leg [Figure 3]. According to the index event, his mental health was normal for his age, and he had no intellectual impairments. Routine tests revealed elevated haemoglobin concentrations The right limb has a level of 5 mg/dL, but no parasites were found on the peripheral smear.8 cm longer than the left on an X-ray of both lower limbs [Figure 4]. Doppler ultrasonography revealed venous malformation in the right leg and thigh, as well as widespread hypertrophy of soft tissue, but no apparent arterial malformation in the lower arm. The patient is currently being closely monitored on a regular basis.
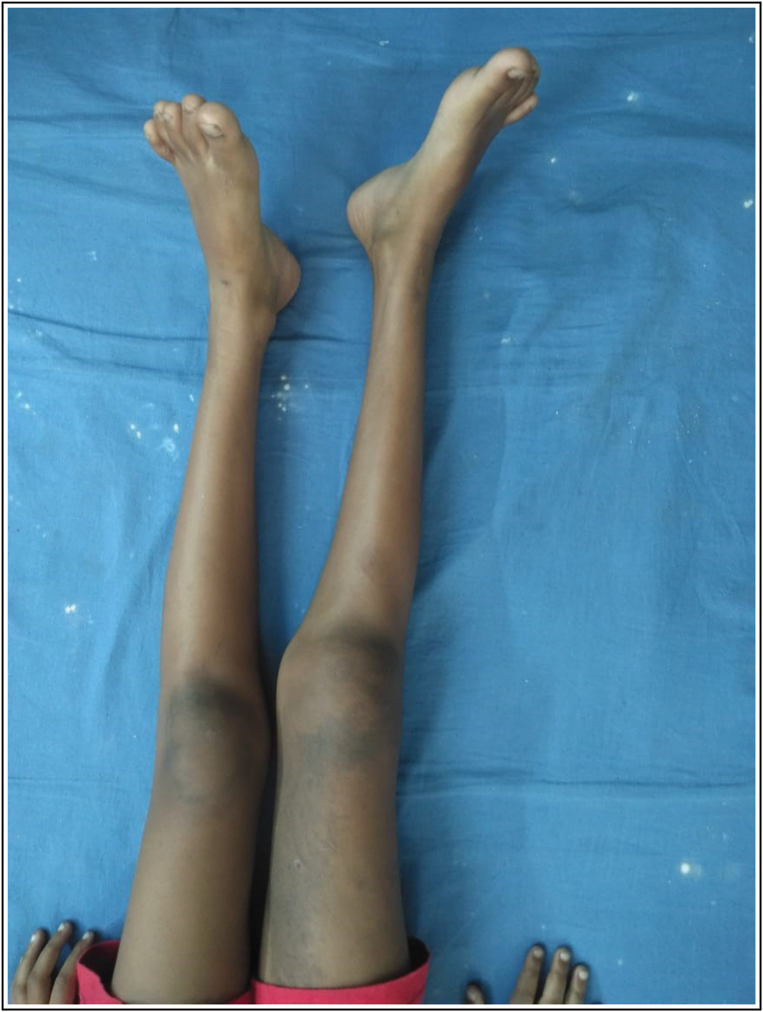
- Clinical Photograph of Right limb
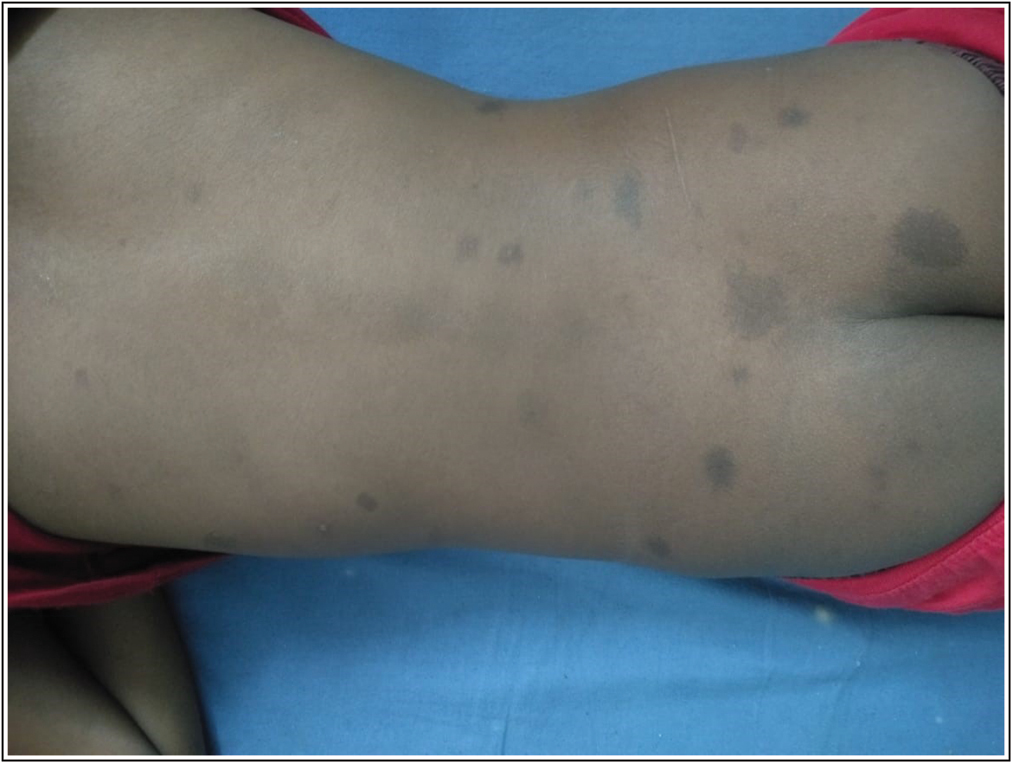
- Photograph of Skin lesion
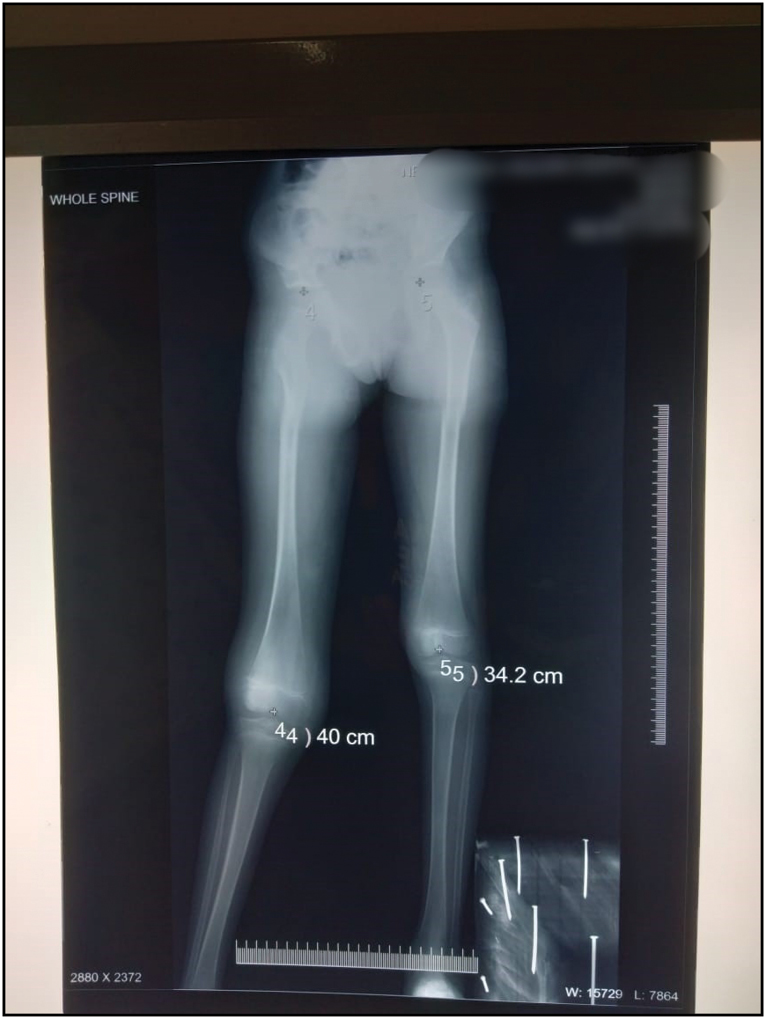
- X ray of patients Showing Lengthen of Femur
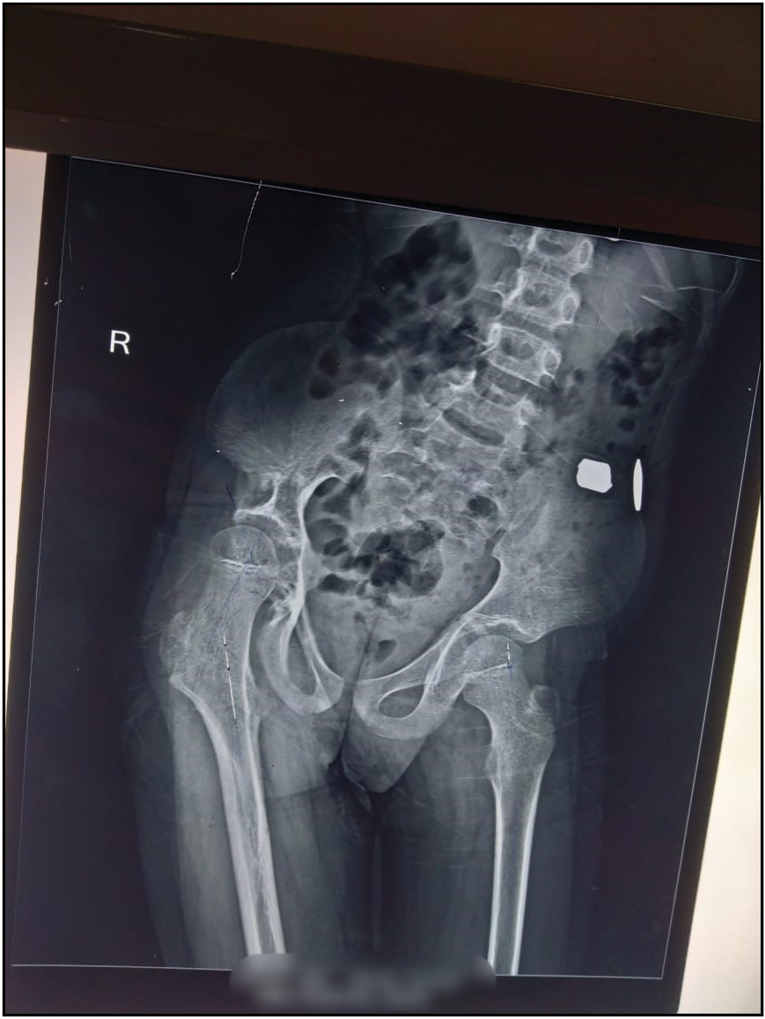
- X-Ray pelvis with both hip
Discussion
The first instance was described in 1900 by Maurice Klippel and Paul Trénaunay in a patient with haemangioma of the epidermis with uneven development of delicate tissue and bone.[1] The cause of KTS is unknown, however several ideas have been proposed, including paradominant inheritance pattern, somatic mosaicism of an ordinarily dominant deadly gene, and soon. disrupted embryonic vasculogenesis and mesodermal abnormalities Although there have been case reports demonstrating different translocations and mutations, but none have been shown to be true. have any kind of clear link to the sickness. Genetic translocation at (8;14) (q22. 3; q13),[2,3] and de novo supernumerary ring chromosome 18,3 are among them. (2q37.3),[4]deletion at the end and 5:11 translocation that is balanced.[5]The number of cases of KTS that have been reported Per 100,000 live births, there are two to five shells.[6,7]Around a century ago, 2 French physicians, Maurice Klippel and Paul Trenaunay, recorded two individuals presenting haemangiomatous lesions of a skin, asymmetrical soft tissue, as well as bone enlargement, as well as coined the term “naevus variqueux osteohypertrophique.”[1] Many years later, Frederick Parks Weber saw a recurrence of identical findings in connection to arteriovenous fistulas. Klippel-Trenaunay-Weber syndrome with an arteriovenous malformation is a variation of Klippel-Trenaunay-Weber syndrome. Throughout history, several review papers of KTS were already published. The incidence and genetic proclivity of this rare illness remain unclear. Capillary abnormalities (port wine stains), larger extremities owing to soft tissue or skeletal hypertrophy, with odd superficial varicosities, notably upon this sides of the body, make up the classic triad. For a patient to also be diagnosed with KTS, at least two of the three cardinal features must be present. Males are more likely than females to develop KTS, with a frequency of 2–5 per 100,000 persons. Usually, only one extremity is affected at a time. Lesions are common from birth, with about 75% of people having them by the age of ten.[8] The aetiology of this disease has been the subject of several theories, which are currently being investigated. Happel argues that the sporadic and familial character of this disease might be explained by the inheritance of a single faulty gene.[9,10] Despite being a rare condition, investigations have shown familial cases of KTS that have not been passed in the Mendelian pattern due to the multifactorial nature of KTS heredity. Skin capillary haemangiomas, bone and soft-tissue hypertrophy, and venous varicosity were the three symptoms when KTS was originally described in 1900.[11]
The illness usually affects only one body region and causes a wide range of clinical symptoms. Hemangioma, the most frequent of the three clinical illness components, is generally the first to show up in patients. Flat hemangioma, often known as port-wine stain, is a birth-visible involution-resistant vascular abnormality. It’s usually one-way and strewn around, seldom crossing the midline. It becomes worse as a child gets older and can affect any area of the body, but it’s most common in the face and cervical region of a child’s anatomy. The lesions may appear bright pink at first, but they will gradually darken over time. This disease can damage bones, muscles, and organs and can be localised or extend deeper into the skin. Hemangiomas have a bad prognosis. Although varicose veins in children with the illness can be visible as early as infancy, they usually become more noticeable as they get older and develop until puberty. The lateral veins are large proximal veins that start in the foot or leg and continue to the gluteus. These areas can cause pain, lymphedema, thrombophlebitis, and ulcers, whether they stay the same or increase over time. Vascular anomalies are believed to be the cause of hypertrophy. Hypertrophy is the third symptom of the disease, which is produced by an increase in bone length and/or circumference due to soft-tissue involvement. It begins at birth and progresses during the first year. The limb will stop developing after the child’s growth cycle is completed in adolescence. Vascular abnormalities of the gastrointestinal and genitourinary systems have been identified, and they can result in significant morbidity and even death.,[12,13,14] There is no treatment for this illness. A majority of these cases are typically diagnosed, but confirmation often necessitates laboratory studies including a relatively high D dimer test, AGGF1 genotype interpretation, as well as radiographic image analysis including such x-rays of both the lower extremities, colour Ultrasound of both the veins and arteries, MRI, MR angiography, as well as catheter angiography. Soft tissue enlargement, as well as the existence of concomitant vascular abnormalities, could be identified in KTS using ultrasonography, Computed tomography, and MRI. Vascular problems with in pelvis and abdomen could also be detected using these scanning technologies. Thrombophlebitis, deep venous thrombosis, pulmonary embolism, and heart problems are among some of the consequences of KTS which may be detected with these diagnostic methods.[15,16] The treatment is often conservative, symptomatic, and medical, with surgical intervention only being necessary in a few rare cases.[17] Regular medical monitoring and radiological surveillance of one of the damaged branches are required for the patient. Several treatment modalities are available and can be used, including[18]: compression stockings for chronic venous insufficiency, intermittent pneumatic compression devices, flash lamp-pumped pulsed dye laser for port-wine stains, surgical correction of varicose veins, and epiphysiodesis for orthopaedic abnormalities to prevent (stop) overgrowing of limbs and bone correction. Compression therapy in the form of an elastic garment or compression bandage has been used as a conservative treatment. This treatment has been shown to help with lymphedema and chronic venous insufficiency. Local wound care, compression dressings, specialised orthopaedic footwear, and lifestyle changes may be advised to help with daily tasks and increase limb utilisation.[19] Vascular therapies for venous insufficiency are usually ineffective due to a high recurrence rate. It really is impossible to ligate or excise huge vascular pools. Foam sclerotherapy assisted by ultrasonography or steam veins sclerosis accompanied with foam sclerotherapy are both less intrusive and hence better options. With encouraging results, the USFS has been promoted as a very successful, minimally invasive, pain-free ambulatory treatment in KTS patients.[20] The treatment objectives are to improve the patient’s condition and treat the consequences of severe lesions. Pulsed dye laser therapy is commonly used to treat port wine stains, which yields better results when applied to lesions on the face and chest rather than the extremities.[21] Nonetheless, it only aids in the treatment of haemangiomas on a superficial level. When varicose veins are present, compression stockings are recommended for venous insufficiency. Only symptomatic superficial varicose veins should be treated surgically. The use of orthopaedic braces to prevent the development of spinal anomalies in instances of lower limb hypertrophy is a feasible option. Corrective bone surgery may be necessary in the future if the limb length differences are significant. The lower leg is the site of malformation in around 95 percent of cases. The anomaly virtually never crosses the midline when it is detected on the trunk. Local hyperaemia and venous stasis generated by the vascular abnormalities cause hypertrophy in both the length and circumference of the afflicted extremities.[22] In this case, however, we discovered extensive capillary abnormalities on both sides of the body, with obvious hypertrophy of the right lower leg. The aetiology of limb atrophy remains unknown. Localized fatty atrophy and vascular atherosclerosis might be to blame in this situation. The urine bladder was extremely large and highly positioned, most likely due to neurofibromatosis, and a soft tissue mass was detected subcutaneously and in the paravertebral area. It is unusual in clinical practise for KTS and neurofibromatosis to coexist.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Identification and molecular characterization of de novo translocation t(8;14)(q22.3;q13) associated with a vascular and tissue overgrowth syndrome. Cytogenet Cell Genet. 2001;95:183-8.
- [CrossRef] [PubMed] [Google Scholar]
- Identification and molecular characterization of a de novo supernumerary ring chromosome 18 in a patient with klippel-trenaunay syndrome. Ann Hum Genet. 2004;68:353-61.
- [CrossRef] [PubMed] [Google Scholar]
- Terminal deletion 2q373 in a patient with klippel-trenaunay-weber syndrome. Fetal Pediatr Pathol. 2013;32:351-6.
- [CrossRef] [PubMed] [Google Scholar]
- Klippel-trenaunay-weber syndrome associated with a 5:11 balanced translocation. Am J Med Genet. 1995;59:492-4.
- [CrossRef] [PubMed] [Google Scholar]
- Naevi and other developmental defects. Vascular naevi. In: Champion RH, Burton JL, Burns DA, Breathnac SM, eds. Textbook of Derma Tology (6th). London: Oxford Blackwell Scientific Publications; 1998. p. :585-8.
- [Google Scholar]
- J Am Acad Dermatol. 2007;56:353-70.:quiz 371-70.
- [CrossRef] [PubMed]
- Vascular malformations, Part Ii: Associated syndromes. J Am Acad Dermatol. 2007;56:541-64.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of an angiogenic factor that when mutated causes susceptibility to klippel-trenaunay syndrome. Nature. 2004;427:640-5.
- [CrossRef] [PubMed] [Google Scholar]
- Hematuria and rectal bleeding in the child with klippel and trenaunay syndrome. Ann Surg. 1976;183:418-28.
- [CrossRef] [PubMed] [Google Scholar]
- Bleeding from cavernous angiomatosis of the rectum in klippel-trenaunay syndrome: Report of three cases and literature review. Am J Gastroenterol. 2001;96:2783-8.
- [CrossRef] [PubMed] [Google Scholar]
- Severe hemorrhage from intestinal hemangiomatosis in klippel-trenaunay syndrome: Pitfalls in diagnosis and management. J Pediatr Gastroenterol Nutr. 1986;5:155-8.
- [CrossRef] [PubMed] [Google Scholar]
- Computed tomography and ultrasound findings in klippel-trenaunay syndrome. J Comput Assist Tomogr. 1983;7:457-60.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular anomalies: What a radiologist needs to know. Pediatr Radiol. 2010;40:895-905.
- [CrossRef] [PubMed] [Google Scholar]
- Klippel and trénaunay’s syndrome, 768 operated cases. Ann Surg. 1985;201:365-73.
- [CrossRef] [PubMed] [Google Scholar]
- prospective study of the impact of laser treatment on vascular lesions. Br J Dermatol. 2000;143:356-9.
- [CrossRef] [PubMed] [Google Scholar]
- Advanced management of venous malformation with ethanol sclerotherapy: Mid-term results. J Vasc Surg. 2003;37:533-8.
- [CrossRef] [PubMed] [Google Scholar]
- Ultrasound-guided foam sclerotherapy in patients with klippel-trenaunay syndrome. Isr Med Assoc J. 2007;9:72-5.
- [Google Scholar]
- Klippel-trénaunay syndrome: Spectrum and management. Mayo Clin Proc. 1998;73:28-36.
- [CrossRef] [PubMed] [Google Scholar]




